Cyclopropanation of Terminal Alkenes via Sequential Atom
Transfer Radical Addition – 1,3-Elimination
Nicholas D. C. Tappin, Weronika Michalska, Simon Rohrbach, Philippe Renaud*
[a]
Abstract: An operationally simple protocol to affect an atom transfer
radical addition (ATRA) of commercially available ICH
2
Bpin to
terminal alkenes has been developed. The intermediate iodide can be
transformed in a one-pot process into the corresponding
cyclopropane upon treatment with tetrabutylammonium fluoride
(TBAF). This method is highly selective for the cyclopropanation of
unactivated terminal alkenes over non-terminal alkenes and electron
deficient alkenes. Due to the mildness of the procedure, a wide range
of functional groups such as esters, amides, alcohols, ketones, and
vinylic cyclopropanes are well tolerated. The whole reaction sequence
relies on simple reagents such dilauroyl peroxide (DLP) and TBAF
and can be run on multi-gram scales in ethyl acetate as a solvent.
Due to their stability and ease of handling, alkylboronic esters are
extremely useful and attractive synthetic intermediates for a broad
range of transformations
[1]
involving carbon–heteroatom
[2]
and
carbon–carbon
[3–6]
bond formation. Boronic esters have also been
involved in a variety of radical reactions.
[7–11]
They are prepared
either via hydroboration,
[12,13]
via reactions of organometallic
species with borate esters and related reagents,
[14]
via C—H
activation process
[15]
and more recently via radical borylation
reactions.
[16]
The nature of the alkyl chain at boron can be easily
modified with a high level of stereocontrol by homologation
reactions upon treatment with carbenoid type reagents.
[17–19]
Our long-standing interest in developing mild methods for
the functionalization of alkenes via radical pathway
[20–32]
incited us
to investigate the iodoalkylation involving a 1-borylated alkyl
radicals leading to γ-iodoalkylboronates. Based on EPR studies
and calculations, Walton, Carboni, and co-workers reported that
1-borylated radicals are stabilized when the boron is sp
2
-
hybridized, however the extent of stabilization is smaller for
radicals substituted by a boronic ester substituent relative to the
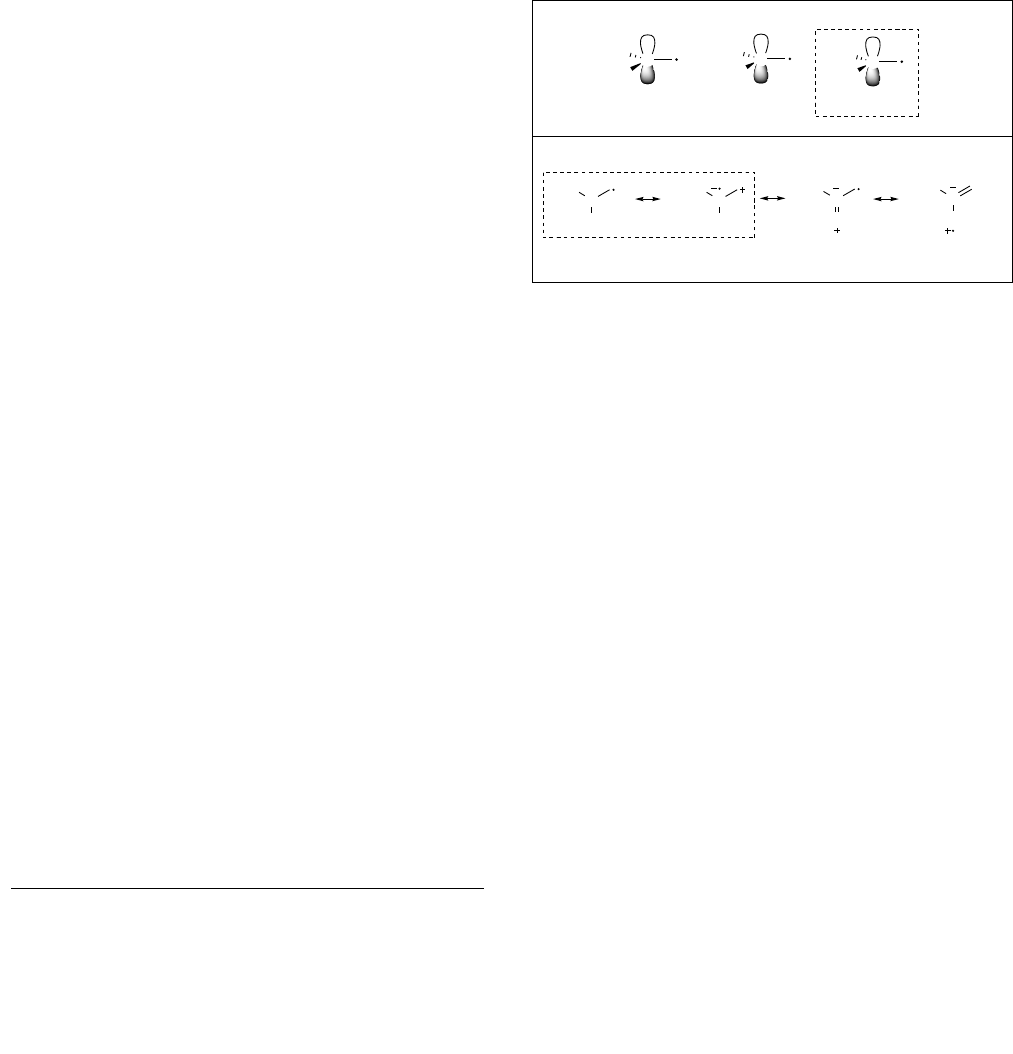
corresponding borinic ester or borane (Figure 1, A).
[33,34]
This
stabilization is the first key feature to design an iodine atom
transfer process according to the pioneering work of Kharasch
and Curran.
[35–41]
Matching the philicities of the radicals and
alkenes involved in the process is the second key feature for the
success of the reaction. Strong polar effects favor an efficient and
selective iodine atom transfer process over an oligomerization
reaction. The resonance hybrids of the 1-borylalkyl radical
suggest that the oxygen lone pairs reduce electrophilicity by the
same mechanism that they decrease radical stabilization (Figure
1 B). Based on these simple considerations, it is expected that
iodine atom transfer between 1-iodoalkylboronic esters and
alkenes should be possible – but still challenging – due to
moderate thermodynamic and polar effects. Recently, Zard and
co-workers rationalized the inefficiency of the xanthate transfer
mediated addition of electrophilic radicals to pinacol vinylboronate
by the lack of favorable thermodynamic and polar effects.
[42–44]
Figure 1. Radical stabilization energies (RSEs) of 1-borylated radicals and
resonance hybrids of boronic esters derived radicals.
[34]
α-Haloboronic esters have been used in tin-mediated (Scheme 1,
C)
[45–47][48]
and metal-catalyzed radical processes.
[49–51]
Chen
reported the Pd(0) catalyzed cyclopropanation of norbornene
using potassium iodomethyltrifluoroborate.
[52]
Interestingly,
pinacol iodomethylboronic ester could also be used for this
transformation (Scheme 1, D). A mechanism involving a
palladocyclobutane intermediate was proposed. Finally, 1-
borylated alkyl radicals were involved in the elegant nickel-
catalyzed alkylation and arylation of α-haloboronic esters
developed by Fu
[53]
and Martin,
[54]
respectively. 1-Borylated alkyl
radicals have also been generated from Barton esters
[55]
and
xanthates
[44]
as well as through radical addition to
vinylboronates.
[42,43,45,56–60]
and to ate complexes of
vinylboronates.
[61–63]
To the best of our knowledge no halogen
atom transfer radical addition (ATRA) process involving 1-
haloalkylboronates have been reported. This reaction would be
highly attractive since it opens a way to prepare in a
straightforward manner 3-haloalkylboronates that are potential
precursors of cyclopropanes via a 1,3-elimination process. Indeed,
Hawthorne and Brown have both reported that 3-
haloalkylboranes are easily converted into cyclopropanes upon
conversion into their ate complexes by treatment with NaOH or
MeLi.
[64–67]
The stable boronic ester was has not yet been used
for this transformation and this attractive 1,3-cyclization has been
limited to a few very simple unfunctionalized cyclopropanes since
the precursors were only available by hydroboration of allylic
halides. We report here, a study of the iodine atom transfer
reactions between pinacol iodomethylboronate 1 and non-
activated alkenes 2 (Scheme 1, E) that led to the development of
a metal-free cyclopropanation procedure demonstrating a high
10.4 kcal mol
–1
8.2 kcal mol
–1
6.7 kcal mol
–1
B
MeO
OMe
B
MeO
OMe
B
MeO
OMe
Decreasing RSEs disfavor ATRA
B
MeO
OMe
Radical electrophilicity favors ATRA
resonance hybrids forms
showing reduced electrophilicity
(disfavor ATRA)
A
B
B
Me
Me
B
MeO
Me
B
MeO
MeO
[a] N. D. C. Tappin, W. Michalska, Dr. S. Rohrbach, Prof. Dr. P.
Renaud
Department of Chemistry and Biochemistry
University of Bern
Freiestrasse 3, CH-3012 Bern, Switzerland
E-mail: [email protected]
Supporting information for this article is given via a link at the end of
the document.

2
chemoselectivity complementary to classical cyclopropanation
methods involving carbenoids, carbenes, and Michael addition
ring closure.
[68]
Scheme 1. Reaction of alkenes with 1-haloalkylboronic esters.
Initial attempts to run the iodine atom transfer reaction
between ICH
2
Bpin 1 and 1-undecene 2a as a substrate
demonstrated that the 3-iodoalkylboronate 3a was decomposing
during purification on silica gel. Eventually, we decided not to
isolate 3a and to convert it immediately into the cyclopropane 4a.
To our delight, preliminary experiments with small amount of
isolated boronic ester 3a showed that the cyclopropane formation
was possible upon treatment with a variety of nucleophiles such
as HO
–
, EtO
–
, and F
–
. Having established that the cyclopropane
formation was possible, we started to optimize the whole process
as outlined in Table 1. Initiation with triethylborane (BEt
3
) and
oxygen or di-tert-butylhyponitrite (DTBHN) were attempted first
using either NaOH or LiOH to trigger the cyclopropanation but
yields remained low (Table 1, entries 1–3). DLP was eventually
identified as a superior initiating system and the use of one
equivalent was necessary to reach the best yields (Table 1,
entries 6–8).
[69]
The reagent used to induce the cyclopropane
formation was found to be less crucial. Good but slightly
irreproducible results were obtained with the biphasic NaOH or
LiOH system (Table 1, entries 3–4), higher yields and
reproducibility were achieved by using LiOEt or TBAF (Table 1,
entries 5, 6). The latter was finally selected for its mildness. The
role of the solvent was found to be less critical. Good yields were
obtained in benzene and chlorobenzene (Table 1, entries 6, 9).
The reaction works also well in DCE (1,2-dichloroethane) (Table
1, entry 10) and it was found later that the reaction gave a similar
yield when run in dry EtOAc (Table 1, entry 11). For the purposes
of this investigation dry benzene was selected as the solvent of
choice but one scale-up experiment was run in EtOAc with a more
polar alkene (see below).
Table 1. Optimization of the cyclopropanation of undecene 2a with pinacol
iodomethylboronate 1.
[a]
Entry
Initiator
Solvent, T
Lewis base
Yield
[b]
1
BEt
3
, air
CH
2
Cl
2
, rt
NaOH
28%
2
BEt
3
, DTBHN
[c]
CH
2
Cl
2
, reflux
NaOH
21%
3
BEt
3
,
[d]
DTBHN
[c]
CH
2
Cl
2
, reflux
LiOH
35%
4
DLP (1 equiv)
C
6
H
6
, reflux
LiOH
71%
5
DLP (1 equiv)
C
6
H
6
, reflux
LiOEt
75%
6
DLP (1 equiv)
C
6
H
6
, reflux
TBAF
73%
7
DLP (0.2 equiv)
C
6
H
6
, reflux
TBAF
40%
8
DLP (1.4 equiv)
C
6
H
6
, reflux
TBAF
71%
9
DLP (1 equiv)
ClC
6
H
5
, reflux
TBAF
74%
10
DLP (1 equiv)
DCE, reflux
TBAF
68%
11
DLP (1 equiv)
EtOAc, reflux
TBAF
74%
[e]
[a] Reactions were run using 2a (1 mmol), 1 (2 mmol) in dry benzene (3.3 mL) then the
nucleophilic solution was added and the reaction mixture stirred vigorously. [b] Yields
determined by GC using pentadecane as internal standard. [c] DTBHN = di-tert-butyl
hyponitrite. [d] Syringe pump addition. [e] NMR yield with 1,4-dimethoxybenzene as
external standard.
The scope of the reaction was investigated with a series of substrates
(Scheme 2). Simple terminal alkenes bearing a wide range of functional
group such as the electron rich aromatic ring of safrole (2c), the silyl ether
2d (using LiOEt instead of TBAF to avoid cleavage of the silyl group), the
ketone 2e, the unprotected primary and secondary alcohols 2f and 2g, the
acylated alcohols 2h and 2i, the benzyl ether 2j, the free carboxylic acid
2k, the esters 2l and 2m, and the secondary and tertiary amides 2o and 2p
gave the desired products in fair to good yields (39–80%). Interestingly, the
cyclopropanation of 2d involved a neo-pentylic iodide showing that the
cyclization mechanism is probably best described as a 1,3-elimination
process in analogy to the reaction of 3-haloalkylboranes where inversion of
the configuration at occurs at the halide and boron substituted carbon
atoms.
[70,71]
When 1,1-disubstituted alkenes were employed (such as 2q),
troubles were encountered with the stability of the intermediate iodides and
a 15% yield was obtained with DLP under thermal initiation. Initiation with
BEt
3
/air turned out to be best since it could be performed at room
temperature to give 4q in 38% yield. Reaction with the
methylenecyclobutane 2r afforded the slightly volatile 4r in a 45% yield.
Following Chen’s work,
[52]
we also cyclopropanated norbornene 2s to 4s in
53% yield. Next, the procedure was used on diverse dienes to test the
chemoselectivity. α,β-Unsaturated esters 2t and 2u reacted selectively at
the unactivated terminal alkene. Acylated prenol, citronellol, nopol, and
cholesterol 2w–2y were selectively cyclopropanated at the terminal non-
activated alkene to give 4w–4y in 48–62% yields. Finally, the
chrysanthemic ester 2z afforded 4z as a single isolated regioisomer without
any modification of the trans/cis 8:2 diastereomeric ratio demonstrating
further the mildness of the reaction conditions.
BpinI
1) initiator
2) F
–
R
+
R
Bu X
Bpin
R
Bpin
OBu
OBu
+
Bu
3
SnH
initiator
71%
C Batey 1996
I Bpin
+
DMF/H
2
O, 90 ºC
Pd(P(t-Bu)
3
)
2
(cat.)
K
2
CO
3
, CsF
D Chen 2014
92% (GC)
E This work
C
9
H
19
solvent
initiator
C
9
H
19
I
pinB
C
9
H
19
Lewis
base
2a
+ ICH
2
Bpin
1
3a
4a

3
Scheme 2. Scope of the cyclopropanation. Procedure: All reactions were run on 1 mmol scale with no special precautions to remove moisture or air. Alkene (1
mmol), ICH
2
Bpin (2 mmol), and DLP (1 mmol) were heated in refluxing benzene (0.3 M) under argon for 4 h; to the cooled reaction mixture was added TBAF solution
(5 equiv, 1.0 M in THF) and stirred for 16 h. Each yield was determined by
1
H-NMR analysis of the crude product mixture after work-up using 1,4-dimethoxybenzene
as reference. a) LiOEt (1.0 M in EtOH) was used instead of TBAF. b) Reaction performed in EtOAc on a 5 mmol scale. c) Initiated with BEt
3
(0.5—1.2 equiv) open
to air, rt. d) Yield estimated by GC analysis using 1,5-cyclooctadiene as reference. See SI for details. e) No erosion of the trans/cis ratio.
As noticed early on, the isolation of the pinacol γ-iodoalkylboronic
esters 3 resulting from the reaction of ICH
2
Bpin 1 over numerous
alkenes was challenging due to their instability during purification
by silica gel chromatography (nonetheless all intermediates, 3a–
z, are characterized). This instability was attributed to C—I bond
labilization by the Lewis-acidic proximal sp
2
-hybridized boron
atom. Since pinanediol boronic esters are known to be
thermodynamically more stable,
[72,73]
we hypothesized that they
may be less Lewis acidic and therefore the pinanediol γ-
iodoalkylboronic esters may be more stable. Satisfyingly, reaction
of the pinanediol iodomethylboronic ester 5 with alkenes 2g
afforded 6g that was stable to silica gel purification in 74%
isolated yield (Scheme 3).
Scheme 3. Isolation of a pinanediol γ-iodoalkylboronic ester.
The mechanism of the cyclopropanation reaction is depicted in
Scheme 4 and corresponds to a classical radical iodine atom
transfer process coupled with a Lewis base promoted 1,3-
elimination (or intramolecular nucleophilic substitution). As
discussed earlier, the iodine atom transfer step is favored by the
higher stabilization of the 1-borylated radical relative to an alkyl
radical. The radical addition to alkenes is controlled by polar and
steric effects: the electron withdrawing boryl group favors the
addition to electron rich alkenes and radical addition are much
faster at the less hindered position. These two effects rationalize
well the observed regioselectitvities reported in Scheme 2.
Scheme 4. Mechanism of the cyclopropanation involving ATRA and 1,3-
elimination processes.
In conclusion, we have reported here an unprecedented approach
for the formation of cyclopropanes relying on a one-pot iodine
atom transfer radical addition followed by an ionic intramolecular
substitution process (1,3-elimination). The whole sequence is
characterized by mild reaction conditions and excellent functional
group compatibility. Interestingly, the high regioselectivity
observed for terminal non-activated alkenes over non-terminal
and electron deficient alkenes cannot be achieved easily using
classical cyclopropanation methods. Finally, the possible isolation
of the intermediate products of iodine atom transfer depicted in
Scheme 3 open new opportunities for further synthetic
applications.
Experimental Section
General cyclopropanation procedure. Reactions were run under argon but
no precaution to eliminate moisture or air was necessary. A solution of
ICH
2
Bpin, 1 (540 mg, 2.0 mmol), DLP (400 mg, 1.0 mmol), and alkene (1.0
O
O
4a 77%
4b 60%
Ph
O
O
4c 39%
O
OH
4e 69%
4f 70%
OTBDMS
4d 58% (LiOEt)
a
OH
OAc
OBz
OBn
CO
2
H CO
2
Et
CO
2
Bn
NHEt
O
NEt
2
O
4g 75%
4h 70%
4i 74%
4n 69%
4m 76% (80%)
b
4k 69%
4j 61%
4o 65%
4p 65%
O
O
4u 56%
4z 56%, trans/cis 8:2
e
O
O
O
O
O
O
4v 54%
O
O
H
H
H
H
O
O
Ph
4t 53%
4y 56%
4x 62%
4w 48%
Alkenes
Dienes
( )
9
( )
9
( )
9
( )
9
( )
9
( )
8
( )
8
( )
8
( )
8
( )
8
( )
8
( )
8
( )
9
( )
9
( )
8
( )
9
( )
8
( )
8
( )
8
1 (2 equiv), DLP (1 equiv)
benzene, reflux, 4 h
then TBAF (5 equiv), rt, 16 h
2a—z
4a—z
R
R
CO
2
Me
4l 71%
( )
8
Ph
4q 15% (38%)
c
CN
4r 45%
c
4s 50%
d
(53%)
c,d
OH
OH
6g 74% (dr 1:1)
( )
9
( )
9
B
O
O
IB
O
O
+
5 (2 equiv)
2g
DLP (1 equiv)
benzene, reflux
I
R
pinB I
R
pinB
pinB R
pinB R
I
pinB I
R
F
2
1
3
F
–
4
ATRA
1,3-elimination
4
mmol) in benzene (3.5 mL) was heated under reflux for 4 h. After cooling
down to rt, a TBAF solution (1.0 M in THF, 5.0 mL, 5.0 mmol) was added
in one portion and the reaction mixture stirred for 16 h. The mixture was
partitioned between TBME and H
2
O, shaken with sat. aq. Na
2
S
2
O
3
and
washed successively with H
2
O, sat. aq. NaHCO
3
and brine. After drying
over Na
2
SO
4
, the residue was filtered through a silica plug to afford the
desired crude cyclopropane.
Acknowledgements
The Swiss National Science Foundation (Project
200020_172621) and the University of Bern are gratefully
acknowledged for financial support.
Keywords: radical reaction • organoboron • cyclopropanation •
alkenes • 1-borylated radical • iodine atom transfer • ATRA •
boronate complexes.
[1] C. Sandford, V. K. Aggarwal, Chem. Commun. 2017, 53, 5481–5494.
[2] T. Chinnusamy, K. Feeney, C. G. Watson, D. Leonori, V. K. Aggarwal, in
Comprehensive Organic Synthesis II (Ed.: P. Knochel), Elsevier,
Amsterdam, 2014, pp. 692–718.
[3] R. Armstrong, V. Aggarwal, Synthesis 2017, 49, 3323–3336.
[4] A. J. J. Lennox, G. C. Lloyd-Jones, Chem. Soc. Rev. 2013, 43, 412–443.
[5] C. Valente, M. G. Organ, in Boronic Acids, Wiley, 2011, pp. 213–262.
[6] A. Bonet, M. Odachowski, D. Leonori, S. Essafi, V. K. Aggarwal, Nat.
Chem. 2014, 6, 584–589.
[7] C. Ollivier, P. Renaud, Chem. Rev. 2001, 101, 3415–3434.
[8] P. Renaud, A. Beauseigneur, A. Brecht-Forster, B. Becattini, V.
Darmency, S. Kandhasamy, F. Montermini, C. Ollivier, P. Panchaud, D.
Pozzi, et al., Pure Appl. Chem. 2007, 79, 223–233.
[9] P. Renaud, in Encyclopedia of Radicals in Chemistry, Biology and
Materials (Eds.: C. Chatgilialoglu, A. Studer), Wiley, Chichester, 2012.
[10] G. Yan, M. Yang, X. Wu, Org. Biomol. Chem. 2013, 11, 7999–8008.
[11] G. Duret, R. Quinlan, P. Bisseret, N. Blanchard, Chem. Sci. 2015, 6,
5366–5382.
[12] A. Pelter, K. Smith, H. C. Brown, Borane Reagents, Academic Press,
London ; San Diego, 1988.
[13] S. J. Geier, C. M. Vogels, S. A. Westcott, in Boron Reagents in Synthesis,
American Chemical Society, 2016, pp. 209–225.
[14] D. G. Hall, in Boronic Acids: Preparation and Applications in Organic
Synthesis and Medicine (Ed.: D.G. Hall), Wiley, 2011, pp. 1–133.
[15] T. Ishiyama, N. Miyaura, in Boronic Acids, Wiley, 2006, pp. 101–121.
[16] G. Yan, D. Huang, X. Wu, Adv. Synth. Catal. 2018, 360, 1040–1053.
[17] D. S. Matteson, J. Org. Chem. 2013, 78, 10009–10023.
[18] D. S. Matteson, in Boron Reagents in Synthesis (Ed.: A. Coca), American
Chemical Society, Washington, 2016, pp. 173–208.
[19] C. G. Watson, P. J. Unsworth, D. Leonori, V. K. Aggarwal, in Lithium
Compounds in Organic Synthesis, Wiley-VCH, Weinheim, 2014, pp.
397–422.
[20] G. Povie, S. R. Suravarapu, M. P. Bircher, M. M. Mojzes, S. Rieder, P.
Renaud, Science Adv. 2018, 4, eaat6031.
[21] D. Meyer, P. Renaud, Angew. Chem. Int. Ed. 2017, 56, 10858–10861.
[22] D. Meyer, E. Vin, B. Wyler, G. Lapointe, P. Renaud, Synlett 2016, 27,
745–748.
[23] G. Povie, A. T. Tran, D. Bonnaffé, J. Habegger, Z. Y. Hu, C. Le Narvor,
P. Renaud, Angew. Chem. Int. Ed. 2014, 53, 3894–3898.
[24] Cao Lidong, Weidner Karin, Renaud Philippe, Adv. Synth. Catal. 2012,
354, 2070–2070.
[25] Cao Lidong, Weidner Karin, Renaud Philippe, Adv. Synth. Catal. 2011,
353, 3467–3472.
[26] K. Weidner, A. Giroult, P. Panchaud, P. Renaud, J. Am. Chem. Soc.
2010, 132, 17511–17515.
[27] A. Beauseigneur, C. Ericsson, P. Renaud, K. Schenk, Org. Lett. 2009,
11, 3778–3781.
[28] A. P. Schaffner, K. Sarkunam, P. Renaud, Helv. Chim. Acta 2006, 89,
2450–2461.
[29] P. Panchaud, P. Renaud, CHIMIA 2004, 58, 232–233.
[30] P. Panchaud, P. Renaud, J. Org. Chem. 2004, 69, 3205–3207.
[31] P. Renaud, P. Renaud, C. Ollivier, P. Panchaud, Angew. Chem. Int. Ed.
2002, 41, 3460–3462.
[32] C. Ollivier, P. Renaud, Angew. Chem. Int. Ed. Engl. 2000, 39, 925–928.
[33] A. McCarroll, J. C. Walton, A. McCarroll, R. Nziengui, B. Carboni, Chem.
Commun. 1997, 2075–2076.
[34] J. C. Walton, A. J. McCarroll, Q. Chen, B. Carboni, R. Nziengui, J. Am.
Chem. Soc. 2000, 122, 5455–5463.
[35] D. P. Curran, in Free Radicals in Synthesis and Biology (Ed.: F. Minisci),
Springer, Dordrecht, 1989, pp. 37–51.
[36] J. Byers, in Radicals in Organic Synthesis, Wiley-Blackwell, 2008, pp.
72–89.
[37] M. S. Kharasch, E. V. Jensen, W. H. Urry, Science 1945, 102, 128–128.
[38] M. S. Kharasch, P. S. Skell, P. Fisher, J. Am. Chem. Soc. 1948, 70,
1055–1059.
[39] D. P. Curran, C. T. Chang, J. Org. Chem. 1989, 54, 3140–3157.
[40] D. P. Curran, M. H. Chen, E. Spletzer, C. M. Seong, C. T. Chang, J. Am.
Chem. Soc. 1989, 111, 8872–8878.
[41] H. Zipse, in Radicals in Synthesis I (Ed.: A. Gansäuer), Springer, Berlin,
2006, pp. 163–189.
[42] H. Lopez-Ruiz, S. Z. Zard, Chem. Commun. 2001, 2618–2619.
[43] M. R. Heinrich, L. A. Sharp, S. Z. Zard, Chem. Commun. 2005, 3077.
[44] B. Quiclet-Sire, S. Z. Zard, J. Am. Chem. Soc. 2015, 137, 6762–6765.
[45] N. Guennouni, F. Lhermitte, S. Cochard, B. Carboni, Tetrahedron 1995,
51, 6999–7018.
[46] R. A. Batey, D. V. Smil, Angew. Chem. Int. Ed. 1999, 38, 1798–1800.
[47] R. A. Batey, D. V. Smil, Tetrahedron Lett. 1999, 40, 9183–9187.
[48] R. A. Batey, B. Pedram, K. Yong, G. Baquer, Tetrahedron Lett. 1996, 37,
6847–6850.
[49] K. Takai, N. Shinomiya, M. Ohta, Synlett 1998, 1998, 253–254.
[50] T. C. Atack, S. P. Cook, J. Am. Chem. Soc. 2016, 138, 6139–6142.
[51] D. Kurandina, M. Parasram, V. Gevorgyan, Angew. Chem. Int. Ed. 2017,
56, 14212–14216.
[52] T. den Hartog, J. M. S. Toro, P. Chen, Org. Lett. 2014, 16, 1100–1103.
[53] J. Schmidt, J. Choi, A. T. Liu, M. Slusarczyk, G. C. Fu, Science 2016,
354, 1265–1269.
[54] S.-Z. Sun, R. Martin, Angew. Chem. Int. Ed. 2018, 57, 3622–3625.
[55] Z. He, P. Trinchera, S. Adachi, J. D. St. Denis, A. K. Yudin, Angew. Chem.
Int. Ed. 2012, 51, 11092–11096.
[56] D. S. Matteson, J. Am. Chem. Soc. 1960, 82, 4228–4233.
[57] D. S. Matteson, A. H. Soloway, D. W. Tomlinson, J. D. Campbell, G. A.
Nixon, J. Med. Chem. 1964, 7, 640–643.
[58] S. A. Green, J. L. M. Matos, A. Yagi, R. A. Shenvi, J. Am. Chem. Soc.
2016, 138, 12779–12782.
[59] A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers, V. K. Aggarwal, Angew.
Chem. Int. Ed. 2018, 57, 2155–2159.
[60] C. Shu, R. S. Mega, B. J. Andreassen, A. Noble, V. K. Aggarwal, Angew.
Chem. Int. Ed. 2018, 57, 15430–15434.
[61] M. Kischkewitz, K. Okamoto, C. Mück-Lichtenfeld, A. Studer, Science
2017, 355, 936–938.
[62] M. Silvi, C. Sandford, V. K. Aggarwal, J. Am. Chem. Soc. 2017, 139,
5736–5739.
[63] N. D. C. Tappin, M. Gnägi-Lux, P. Renaud, Chem. Eur. J. 2018, 24,
11498–11502.
[64] M. F. Hawthorne, J. A. Dupont, J. Am. Chem. Soc. 1958, 80, 5830–5832.
[65] M. F. Hawthorne, J. Am. Chem. Soc. 1960, 82, 1886–1888.
[66] H. C. Brown, S. P. Rhodes, J. Am. Chem. Soc. 1969, 91, 2149–2150.
[67] H. C. Brown, S. P. Rhodes, J. Am. Chem. Soc. 1969, 91, 4306–4307.
[68] H. Lebel, J.-F. Marcoux, C. Molinaro, A. B. Charette, Chem. Rev. 2003,
103, 977–1050.
[69] C. Ollivier, T. Bark, P. Renaud, Synthesis 2000, 1598–1602.
[70] H. L. Goering, S. L. Trenbeath, J. Am. Chem. Soc. 1976, 98, 5016–5017.
[71] M. E. Gurskii, T. V. Potapova, K. L. Cherkasova, Yu. N. Bubnov, Russ.
Chem. Bull. 1988, 37, 334–338.
[72] C. D. Roy, H. C. Brown, Monatsh. Chem. 2007, 138, 879–887.
[73] C. D. Roy, H. C. Brown, J. Organomet. Chem. 2007, 692, 784–790.
